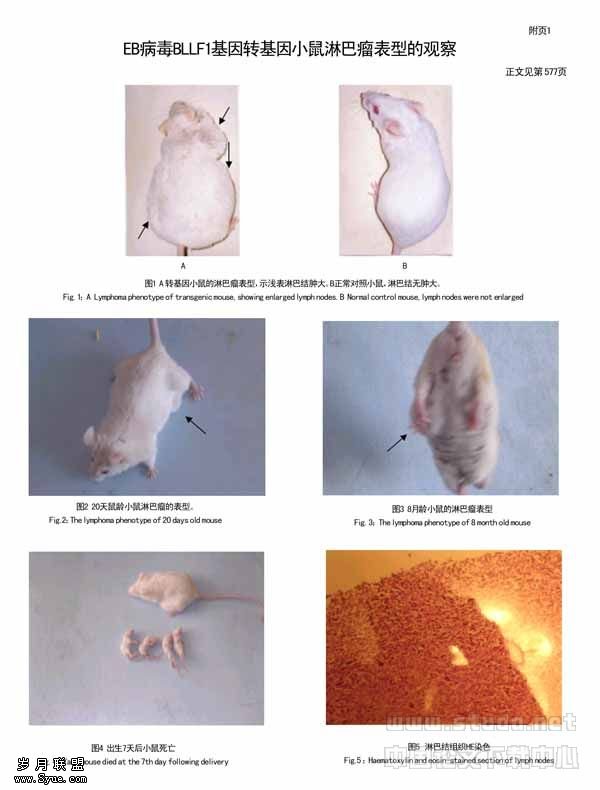
分泌型单链双功能抗体分子间接头的设计及软件分析
【关键词】 抗体,双特异性;肿瘤坏死因子类;生物学
Design and analysis of linkers among secretary singlechain bifunctional antibody fusion molecules with bioinformatics software
【Abstract】 AIM: AIM: To analyze the secondary structure and the physical and chemical characteristics of the secretary antiosteosarcoma singlechain bifunctional antibody fusion protein with bioinformatics software. METHODS: The sites of enzyme were selected for the sequence of linker. Bioinformatics software was used to analyze structural change of the singlechain antibody fragments of variable domaintumor necrosis factor (ScFvTNF) fusion protein, and then DNAssist and ANTHEPROT V5 were used to analyze its sequence, secondary structure, and physical and chemical characteristics. RESULTS: On the fusion genes, the sites of enzyme were selected for the sequence of linker, and the secondary structure of the fusion protein was obtained with DNAssist, and the physical and chemical characteristics of the fusion protein were forecasted with ANTHEPROT V5. CONCLUSION: The software for bioinformatics should be fully utilized to analyze biological information, and be improved continuously. Bioinformatics can shorten the course of pharmaceutical development, at the same time can help us to explore the genebased medicine.
【Keywords】 antibodies, bispecific; tumor necrosis factors; computational biology
【摘要】 目的: 利用生物信息学软件分析分泌型抗骨肉瘤单链双功能抗体融合蛋白的二级结构及其理化性质. 方法: 构建融合蛋白时,先进行连接肽的设计,选用通用酶切位点序列,运用DNA分析软件(DNAssist)和蛋白质分析软件(ANTHEPROT V5)分析预测融合蛋白的氨基酸序列、二级结构及其理化性质. 结果: 在编码ScFv与TNF的碱基之间引入连接肽,通过酶切位点获得的DNA序列,运用DNAssist核酸序列软件获得了融合蛋白的二级结构,并运用蛋白质分析软件(ANTHEPROT V5)预测了融合蛋白的理化性质. 结论: 应该充分利用生物信息学软件分析生物学信息,并不断对软件本身进行改进,生物信息学软件可提高药物开发进程,可利用生物信息学和遗传学信息来寻找和开发以基因为基础的药物.
【关键词】 抗体,双特异性;肿瘤坏死因子类;计算生物学
0引言
人类基因组计划的目的之一在于阐明人体3~4万种蛋白质的结构、功能、相互作用以及与人类各种疾病之间的关系和各种预防方法,还包括药物在内的治疗方法,即生物大分子结构模拟和药物设计[1-4]. 重组融合蛋白是通过DNA重组的方法,将功能上相关的两种蛋白用连接肽连接,以达到优化蛋白功能的目的,如免疫毒素和细胞因子融合蛋白,并已用于肿瘤治疗[5]. 融合蛋白的构建中,连接肽的选择对保留每种成分的活性有重要意义[6]. 为了鉴定重组导向分子ScFvTNF的序列及其二级结构,通过重组PCR方法在编码ScFv与TNF的碱基之间引入酶切位点,并克隆到逆转录病毒表达载体PLxSN上表达,并运用生物信息学资源进行软件分析.
1材料和方法
1.1材料联想昭阳K71;奔腾4处理器;内存256M;操作系统WindowsXP. DNA分析软件(DNAssist)和蛋白质分析软件(ANTHEPROT V5)的下载地址为和.
1.2方法在不考虑重组蛋白质的折叠的情况下,通常采用酶切位点直接连接,实验方便,容易鉴定. 为保证蛋白质的活性,保持蛋白质的自然折叠是非常重要的. 在融合蛋白的构建中,连接肽的选择对保留每种成分的活性有重要意义. 连接肽应选用非极性的疏水氨基酸,长度在10~15个氨基酸之间,能使两种分子形成保持活性必需的空间构象. 我们在构建融合蛋白时,为了提高重组蛋白质分子ScFvTNF的活性,可选用通用连接肽即甘氨酸和丝氨酸的重复序列.
通过基因重组技术在编码ScFv与TNF的碱基之间引入编码KLGGKLGGKLGG连接肽,其碱基序列为:
AAACTTGGGGGGAAACTTGGGGGGAAACTTGGGGGG.
分析序列:ScFvTNF
5′GCGAATTCATGGAATGGAGCGGGGTCTTTCTCTTCCTCCTGTCAATAATTGCAGGTGTCCATTGCCAGGTCCAGC
TGCAGCAGTCTGGACCTGAGCTGGTGAGGCCTGGGACTTCAGTGAGGATATCCTGCAAGGCTTCTGGCTACACCTT
CACAATTTACTATATAAACTGGGTGAAGCAGAGGCCTGGACAGGGACTTGAGTGGATTGGATGGATTTATCCTGGA
AATTTTAATACTAAGTACAATGAGAAGTTCAAGGGCAAGGCCACACTGACTGCGACAAATCCTCCAGCACAGCCT
ACATGCAGCTCAGCAGCCTGACCTCTGAGGATTCTGCGGTCTATTTCTGTGCAAGGGGGGGAAACCTGGACTACTT
TGACTACTGGGGCCAAGGCGCCACTCTCACAGTCTCCTCAGCCAAAACGACACCCCCACCCGTCTATCCCTTGGCC
CAAACTTGGGGGGAAACTTGGGGGGAAACTTGGGGGGGTCAGATCATCTTCTCGAACCCCGAGTGACAAGCCTGTAGC
CCATGTTGTAGCAAACCCTCAAGCTGAGGGGCAGCTCCAGTGGCTGAACCGCCGGGCCAATGCCCTCCTGGCCAAT
GGCGTGGAGCTGAGAGATAACCAGCTGGTGGTGCCATCAGAGGGCCTGTACCTCATCTACTCCCAGGTCCTCTTCA
AGGGCCAAGGCTGCCCCTCCACCCATGTGCTCCTCACCCACACCATCAGCCGCATCGCCGTCTCCTACCAGACCAA
GGTCAACCTCCTCTCTGCCATCAAGAGCCCCTGCCAGAGGGAGACCCCAGAGGGGGCTGAGGCCAAGCCCTGGTA
TGAGCCCATCTATCTGGGAGGGGTCTTCCAGCTGGAGAAGGGTGACCGACTCAGCGCTGAGATCAATCGGCCCGA
CTATCTCGACTTTGCCGAGTCTGGGCAGGTCTACTTTGGGATCATTGCCCTGTAAGGATCCGC3′
2结果
2.1连接肽间接连接获得ScFvTNF DNA序列通过DNA分析软件DNAssist分析ScFvTNF序列的结果.
Translate (DNA翻译蛋白质):
>UNKNOWN
MEWSGVFLFLLSIIAGVHCQVQLQQSGPELVRPGTSVRISCKASGYTFTIYYINWVKQRPGQGLEWIGWIYPGNFNTK
YNEKFKGKATLTATNPPAQPTCSSAAPLRILRSISVQGGETWTTLTTGAKAPLSQSPQPKRHPHPSIPWPKLGGKLGGK
LGGVRSSSRTPSDKPVAHVVANPQAEGQLQWLNRRANALLANGVELRDNQLVVPSEGLYLIYSQVLFKGQGCPSTHV
LLTHTISRIAVSYQTKVNLLSAIKSPCQRETPEGAEAKPWYEPIYLGGVFQLEKGDRLSAEINRPDYLDFAESGQVYFG
IIAL
Find ORF (分析连接肽间接连接ScFvTNF序列的开放阅读框架):
ORF1: 5′ATGGAATGGAGCGGGGTCTTTCTCTTCCTCCTGTCAATAATTGCAGGTGTCCATTGCCAGGTCCAGCTG
CAGCAGTCTGGACCTGAGCTGGTGAGGCCTGGGACTTCAGTGAGGATATCCTGCAAGGCTTCTGGCTACACCTTCA
CAATTTACTATATAAACTGGGTGAAGCAGAGGCCTGGACAGGGACTTGAGTGGATTGGATGGATTTATCCTGGAAA
TTTTAATACTAAGTACAATGAGAAGTTCAAGGGCAAGGCCACACTGACTGCGACAAATCCTCCAGCACAGCCTACA
TGCAGCTCAGCAGCC3′
ORF2: 5′ATGGAGCGGGGTCTTTCTCTTCCTCCTGTCAATAATTGCAGGTGTCCATTGCCAGGTCCAGCTGCAGCAG
TCTGGACC3′
ORF3: 5′ATGGATTTATCCTGGAAATTT3′
ORF4:5′ATGAGAAGTTCAAGGGCAAGGCCACAC3′
ORF5:5′ ATGCAGCTCAGCAGCCTGACCTCTGAGGATTCTGCGGTCTATTTCTGTGCAAGGGGGGGAAACCTGGAC
TACTTTGACTACTGGGGCCAAGGCGCCACTCTCACAGTCTCCTCAGCCAAAACGACACCCCCACCCGTCTATCCCTT
GGCCCAAACTTGGGGGGAAACTTGGGGGGAAACTTGGGGGGGTCAGATCATCTTCTCGAACCCCGAG3′
ORF6:5′ATGTTG3′
ORF7:5′ATGCCCTCCTGGCCAATGGCGTGGAGC3′
ORF8:5′ATGGCGTGGAGC3′
ORF9:5′ATGTGCTCCTCACCCACACCATCAGCCGCATCGCCGTCTCCTACCAGACCAAGGTCAACCTCCTCTCTGC
CATCAAGAGCCCCTGCCAGAGGGAGACCCCAGAGGGGGCTGAGGCCAAGCCCTGGTATGAGCCCATCTATCTGGG
AGGGGTCTTCCAGCTGGAGAAGGGTGACCGACTCAGCGCTGAGATCAATCGGCCCGACTATCTCGACTTTGCCGAG
TCTGGGCAGGTCTACTTTGGGATCATTGCCCTG3′
ORF10:5′ATGAGCCCATCTATCTGGGAGGGGTCTTCCAGCTGGAGAAGGGTGACCGACTCAGCGCTGAGATCAAT
CGGCCCGACTATCTCGACTTTGCCGAGTCTGGGCAGGTCTACTTTGGGATCATTGCCCTG3′
2.2连接肽间接连接获得DNA序列ScFvTNF通过蛋白质分析软件ANTHEPROT V5的分析表明,经连接肽间接连接不同基因融合表达蛋白ScFvTNF二级结构的分析结果(图1),经连接肽间接连接不同基因融合表达蛋白ScFvTNF的理化性质的分析结果(图2),经连接肽间接连接不同基因融合表达蛋白ScFvTNF潜在的信号肽与断裂位点的分析结果(图3).
H代表螺旋,窗口中显示为蓝色;E代表折叠,窗口显示为橙色;T代表转角,窗口显示为绿色;C代表其他松散结构,窗口显示为黑色.
图1ScFvTNF蛋白质的二级结构(略)
图2ScFvTNF蛋白质的理化性质(略)
图3ScFvTNF蛋白质的潜在的信号肽与断裂位点(略)
3讨论
传统生物学认为,蛋白质的序列决定了它的结构,也就决定了它的功能[7-9]. 因此,随着近10 a来生物学分子序列信息的爆炸性增长,大大促进了各种序列分析和预测技术的,目前已经可以用理论预测的方法获得大量的结构和功能信息,用生物信息学的方法,通过机模拟和计算来“预测”出未知蛋白质信息或提供与之相关的辅助信息,可以用较低的成本和较快的时间就能获得可靠的结果[9-10]. 但由于蛋白质复杂的结构及其影响因素的多样性,用一种方法预测结果往往不准,需要用多种方法综合比较分析,以求得最佳结果.DNA序列分析软件DNAssist和蛋白质分析软件(ANTHEPROT V5)是生物信息学领域不可多得的综合性软件.
基于生物大分子结构的药物设计是生物信息学中极为重要的研究领域,为了抑制某些酶和蛋白质的活性,在已知其三级结构的基础上,可以利用分子对接算法,在计算机上设计抑制分子,作为候选药物. 从方法上看,有演绎法和归纳法两种途径. 前者主要是从一些基本原理或假设出发来预测和研究蛋白质的结构和折叠过程,分子力学和分子动力学属这一范畴. 后这主要是从观察和已知结构的蛋白质结构出发来预测未知蛋白质的结构,同源模建和Threading法属于这一范畴. 该项技术算法十分复杂,尚未成熟,虽然经过30余年的努力,蛋白质结构预测研究现状仍远远不能满足实际需要,PDB及MMDB数据库目前仍然不收录软件预测出来的蛋白质高级结构模型[7-8].
重组融合蛋白是通过DNA重组的方法,将功能上相关的两种蛋白用连接肽连接,以达到优化蛋白功能的目的,如免疫毒素和细胞因子融合蛋白,并已用于肿瘤. 在融合蛋白的构建中,连接肽的选择对保留每种成分的活性有重要意义. 连接肽应选用非极性的疏水氨基酸,长度在10~15个氨基酸之间,能使两种分子形成保持活性必需的空间构象. 为了分析重组蛋白质分子ScFvTNF的活性,通过重组PCR方法在编码ScFv与TNF的碱基之间引入酶切位点,与在编码ScFv和TNF的碱基之间引入编码KLGGKLGGKLGG连接肽的碱基序列,两种方法获得的DNA序列,并运用DNAssist核酸序列分析软件分析ScFvTNF基因序列翻译并获得了氨基酸序列及其开放性阅读框架,以及运用蛋白质分析软件(ANTHEPROT V5)分析融合蛋白的二级结构,分析融合蛋白的理化性质及其潜在的信号肽与断裂位点. 分析结果表明,两种方法获得的DNA序列的融合蛋白质的二级结构,理化性质及其潜在的信号肽与断裂位点的信息未显示差异.
【】
[1] Chen T, Jaffe JD, Church GM. Algorithms for identifying protein crosslinks via tandem mass spectrometry[J]. J Comput Biol, 2001,8(6):571-583.
[2] George RA, Heringa J. SnapDRAGON: A method to delineate protein structural domains from sequence data[J]. J Mol Biol, 2002,316(3):839-851.
[3] Zhu JQ, Zhang CW, Rao Z, et al. Biochemical and biophysical analysis of heptad repeat regions from the fusion protein of Menangle virus, a newly emergent paramyxovirus[J]. Arch Virol, 2003,148(7):1301-1316.
[4] Jinshu X, Jingjing L, Duan P, et al. The immunogenicity of recombinant and dimeric gonadotrophinreleasing hormone vaccines incorporating a Thelper epitope and GnRH or repeated GnRH units[J]. J Immunol Methods, 2004,289(12):111-122.
[5] Zheng LY, Xi YZ, Kong FH, et al. Molecular design and construction of IL6D24PE40KDEL, a novel recombinant interleukin6pseudomonas exotoxin fusion protein, having targeted cytotoxicity for leukemias expressing interleukin6 receptors[J]. Chin Med J (Engl), 2003,83(14):1246-1250.
[6] Wu AM, Yazaki PJ. Designer genes: Recombinant antibody fragments for biological imaging[J]. Q J Nucl Med, 2000,44(3):268-283.
[7] Peitsch MC, Wilkins MR, Tonella L, et al. Large scale protein modelling and integration with the SWISS PROT and SWISS2DPAGE databases: The example of Escherichia coli[J]. Electrophoresis, 1997,18(34):498-501.
[8] Guex N, Peitsch MC. SWISSMODEL and the SwissPdbViewer:An environment for comparative protein modelling[J]. Electrophoresis, 1997,18(15): 2714-2723.
[9] Guex N, DiemandA, Peitsch MC. Protein modelling for all[J]. Trends Biochem Sci, 1999,24(9): 364-367.
[10] Guigo R. Computational gene identification[J]. J Mol Med, 1997,75(6):389-393.