人类胚胎肌肉细胞体外自发恶性转化抑癌基因的改变
【摘要】 目的: 研究人类胚胎肌肉源性自发恶性转化细胞系抑癌基因的改变.方法: 运用免疫组化、PCR,PCR产物直接测序研究该类细胞系抑癌基因p16,p15,MTAP(甲硫氨酸磷酸化酶)和p53基因及蛋白的改变.结果: 在染色体9p21处存在纯合性片段缺失,包括p16,p15及MTAP;对p53基因突变率高发的外显子7,8及内含子7测序发现存在范围较大的突变,外显子突变率高达13%,皆为点突变,而内含子变异更甚,包括小片段缺失、插入、移位及点突变,其几与正常p53序列无明显相似性,在测序的6个细胞系中(含5个不同遗传背景)突变序列具有高度一致性.结论: 多种抑癌基因的变异在正常胚胎肌肉细胞自发恶性转化进程中具有重要的作用.
【关键词】 恶性转化 基因 肿瘤抑制 肉瘤 染色体9p21 p53基因 突变
0引言
抑癌基因的失活或突变产物会对细胞周期增殖调控和细胞凋亡产生影响,干扰其正常周期过程,为细胞恶性转化提供潜在的基因基础.其中,比较公认的几个对细胞恶性转化影响较大的基因有周期素依赖性蛋白激酶抑制家族CDKI、Rb和p53等[1,2].目前,体外培养人类细胞自发恶性转化的报道很少[3-7],近期的研究发现,体外长期培养的间充质干细胞会发生恶性转化,其机制不清,可能与癌基因的激活和抑癌基因的失活有关,研究发现p16基因的失活和c?myc基因扩增在其中发挥作用[4-5].我们通过正常人类胚胎肌肉细胞培养获得能稳定传代的自发恶性转化细胞系,该类细胞系通过免疫组化及病理研究发现其体内可表现为纤维肉瘤、软骨肉瘤及横纹肌肉瘤的特性(前期研究数据证实)[8].我们对6个胚胎自发恶性转化细胞系(5个不同遗传背景)的研究发现,其在9p21处存在片段缺失.在对p53基因的分析上,对其突变频率较高的外显子 7,8测序发现突变率高达13%,而内含子7更与正常序列无明显相似性. 且在测序的6个样本中具有具有高度的一致性,提示其可能存在一共同的突变机制及较强的选择优势.
1材料和方法
1.1细胞培养人类胚胎肌肉细胞原代培养和传代方法[8].在取材5个不同遗传背景胚胎肌肉进行培养4~7 mo后自发恶性转化,并能稳定维持体外长期传代,至今恶性转化的细胞体外培养已达1-3 a.其中有2个细胞系来自同一个胚胎肌肉细胞的不同培养时期恶性转化,命名为MS5B和MS5C.其它4个细胞系分别命名为MS0812, MS0504, MS3, MS4. MS0812和MS0504细胞系的性质在我们的前期研究中已经有报道[8].
1.2基因组DNA的提取采用宝生物公司的Universal Genomic DNA Extraction Kit Ver. 3.0(TaKaRa Biotechnology (Dalian)Co., Ltd.),按使用说明规范操作.
1.3PCR扩增缺失分析对6个细胞系的p16基因外显子1,2,3,p15基因外显子1及MTAP外显子2~7进行扩增,阳性对照为正常胚胎肌肉细胞.阳性模板质控为β?actin.50×反应体系,10 mmol/L Tris(pH 8.6), 50 mmol/L KCl, 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 0.4 mmol/L 引物,50 ng DNA模板, 5单位Taq聚合酶(TaKaRa TaqTM, TaKaRa Biotechnology Co.,Ltd. ).反应条件及引物序列见表1.产物于20 g/L琼脂糖上电泳后置紫外分析仪照相.
表1引物序列(略)
1.4p53外显子(7-8)测序对6个胚胎自发恶性转化肌肉细胞系(MS0812, MS0504,MS3,MS4,MS5B,MS5C)提取DNA, 扩增p53外显子7,8,PCR产物送上海生工公司纯化后直接测序[9],测序仪为ABI PRISM 3730,测序染料为BigDye terminator.
1.5p16免疫组化将肿瘤细胞接种于35 mm 培养皿,待细胞贴壁饱和度80%左右,弃去培养基,按单层贴壁细胞免疫组化实验步骤操作,一抗为20倍稀释的鼠抗p16 mAb(北京中衫金桥生物技术有限公司),应用AEC染色法进行显色后照相,阳性对照用人正常胚胎肌肉细胞.
2结果
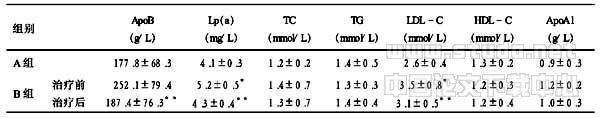
2.1肿瘤细胞系9p21处片段缺失在6个样本的PCR扩增中都出现p16外显子1,2,3,p15外显子1及MTAP外显子4,5,6,7的纯合性缺失(图1A-D).而MTAP外显子2,3则有模糊条带及非特异性扩增(图未示),正常对照则无非特异性扩增(图1E),提示其可能在附近存在断裂位点.
A:p16外显子1纯合性缺失(B,C和D模板顺序与A相同); B:p16外显子2纯合性缺失; C:p16外显子3纯合性缺失;D:p15外显子1纯合性缺失;E:正常肌肉细胞MTAP外显子2~7; F: 各细胞系质控β?actin. M:marker: 1: 正常胚胎肌肉细胞; 2: MS0812; 3: MS0504; 4: MS3; 5: MS4; 6: MS5B; 7: MS5C.
图1肿瘤细胞系9p21处片段缺失(略)
2.2p16免疫组化阳性对照正常胚胎肌肉细胞可见胞质泛染(图2A),而肉瘤细胞系则显示阴性(图2B).
A:正常胚胎肌肉细胞阳性; B:肉瘤细胞系.
图2p16免疫组化阴性(MS3细胞系)(略)
2.3p53外显子7,8及内含子7测序在对6个样本(MS0812, MS0504,MS3,MS4,MS5B,MS5C)的p53外显子7-8测序发现其外显子7与正常p53序列比对其同源性为92%,外显子8为86%左右,内含子7与正常p53序列比对部分几无明显相似性,予人工比对,其余部分可比对(图3).其中,外显子皆为点突变,内含子突变类型有点突变,缺失、插入.
3讨论
p16蛋白能抑制cyclin D/cdk4,阻止pRb磷酸化,从而抑制细胞分裂进入S期,在多种肿瘤中可观察到其失活[10-12].我们通过对人胚胎肌肉细胞的长期培养,约4~7 mo后其自发转化为恶性增殖细胞系[8],PCR及免疫组化结果均支持其存在缺失,而非单纯的点突变.
MTAP蛋白是嘌呤和甲硫氨酸合成补救途径中一个关键酶[13],在人或鼠源的多种恶性细胞系和人多种肿瘤中有较高频率的纯合性缺失,多与p16缺失伴随出现[10].报道在96个骨肉瘤组织中至少一个外显子缺失率的达到37.5%[12].我们对人胚胎肌肉恶性化细胞系体内研究发现该类细胞系表现肉瘤性质[8].PCR扩增其MTAP外显子2~7结果显示4~7纯合性缺失.
p53蛋白在超过半数以上的不同肿瘤中存在突变,其中错义突变约占74%,高于其它抑癌基因发生的频率,从而导致其正常功能的失活及突变蛋白的产生[14].p53基因的突变在软组织肉瘤中很常见,特别是在转移组织及高度恶性化的类型中[15].而且,研究发现各种p53突变对肿瘤的恶性化程度、肉瘤患者的生存率等有显著的影响,即野生型p53功能的消失或抑制,突变型p53蛋白的获得性致癌能力会促进肿瘤的发生,降低患者生存率[14].所以,对p53基因的研究有助于更好的阐明细胞自发恶性化进程的分子机制.而文献报道关于肉瘤p53基因的变异大多是点突变(包括内含子)[16]引起的蛋白编码改变,从而产生突变型p53蛋白.现已在GenBank数据库中确认的关于p53的SNP位点很多,我们在6个细胞系(5个不同遗传背景自发恶性化)的12次测序结果(双向测序)显示,其外显子突变与现有确认的p53 SNP位点是不同的;内含子7更表现出极大的基因变异,其中包括小片段缺失及点突变,这与以往报道的p53内含子突变是完全不同的[17],且12次测序结果具有高度一致性.
A: 上序列为肿瘤细胞系(6个细胞系测序序列相同)序列,下序列为正常人p53外显子7; B:测序p53内含子7上半部分序列, 因其在GenBank数据库比对后示无明显相似序列,予人工比对; C:测序p53内含子7下半部分序列, 使用BLAST比对; D:测序p53外显子8.
图3测序结果 (略)
综上研究结果,我们初步发现在人类胚胎肌肉自发恶性转化细胞系中一些抑癌基因的改变:①9p21处存在片段缺失,其中包括p16、p15及MTAP.其缺失导致周期蛋白抑制功能的丧失,在基因水平上为肿瘤细胞失控的分裂增殖能力解除了限制,分裂能力的增强将加速基因突变频率及癌基因的累积.②p53抑癌基因在外显子7、8的点突变及内含子的大范围改变,将肯定影响p53蛋白的转录及表达[14,18], 3. 6个自发恶性化细胞系结果完全一致,提示其突变模式具有强大的选择优势.
然而,我们对胚胎肌肉来源的自发恶性转化肉瘤细胞系CDKI家族及p53基因的研究还只局限在基因组水平,且还有其他突变率较高的区域未予明确.故接下来仍须在CDKI家族及p53通路等方面进行更深入的研究.相信随着对自发恶性化肉瘤细胞系抑癌基因的深入研究,将有助于更好的解释人正常胚胎肌肉细胞自发恶性化进程的关键分子学机制,为寻找新的基因途径提供帮助.
【文献】
[1] Wiman KG. The retinoblastoma gene: role in cell cycle control and cell differentiation [J]. FASEB J, 1993, 7(10):841-845.
[2] Li Y, Nichols MA, Shay JW, et al. Transcriptional repression of the D?type cyclin?dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb [J]. Cancer Res, 1994, 54(23):6078-6082.
[3] Xu W, Qian H, Zhu W, et al. A novel tumor cell line cloned from mutated human embryonic bone marrow mesenchymal stem cells [J]. Oncol Rep, 2004, 12(3):501-508.
[4] Masako M, Yasuo M, Hesed M, et al. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation [J]. Stem Cells, 2006, 24(4):1095-1103.
[5] Daniel R, Javier GC, Mar?a C, et al. Spontaneous human adult stem cell transformation [J]. Cancer Res, 2005, 65(8): 3035-3039.
[6] Gibbs CP, Kukekov VG, Reith JD,et al. Stem?like cells in bone sarcomas: implications for tumorigenesis [J]. Neoplasia, 2005, 7(11):967-976.
[7] 程天民,史春梦,粟永萍, 等. 成体干细胞在体外培养扩增中的自发恶性转化 [J].中华医学杂志, 2005, 85(27): 1883-1884.
[8] 杨立业,郑佳坤,陈强, 等. 一种新的肉瘤细胞系及其建立方法和应用[P].[申请(专利)]]:200610036856,[公开(公告)]:1904037. Http://www.cnpatent.com/ zljs.asp.
[9] Kretz KA, Carson GS, O?Brien JS. Direct sequencing from low?melt agarose with sequenase [J]. Nucleic Acids Res, 1989, 17(14):5864.
[10] Kuerbitz SJ, Malandro J, Graff JR, et al. Deletion of p16INK4A/CDKN2 and p15INK4B in human somatic cell hybrids and hybrid?derived tumors [J]. Cell Growth Differ, 1999, 10(1):27-33.
[11] Nielsen GP, Burns KL, Rosenberg AE, et al. CDKN2A gene deletions and loss of p16 expression occur in steosarcomas that lack RB alterations [J]. Am J Pathol, 1998,153(1): 159-163.
[12] Kovar H, Jug G, Aryee DN, et al. Among genes involved in the RB dependent cell cycle regulatory cascade, the p16 tumor suppressor gene is frequently lost in the Ewing family of tumors [J]. Oncogene, 1997, 15(18): 2225-2232.
[13] Schlenk F. Methylthioadenosine// Advances in enzymology and related areas in molecular biology [M]. New York: John Wiley, 1988, 2(61):195-265.
[14] van Oijen MG, Slootweg PJ. Gain?of?function mutations in the tumor suppressor gene p53 [J]. Clin Cancer Res, 2000, 6(6):2138-2145.