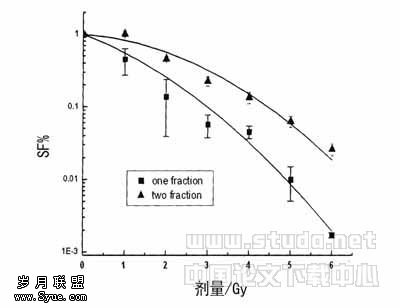
Alport综合征基因学发病机制研究进展
Alport综合征(Alport syndrome,AS),又称为遗传性肾炎、家族性肾炎、遗传性进行性肾炎,是一种主要表现为血尿、肾功能进行性减退、神经性耳聋和眼部异常的遗传性肾小球基底膜( glomerular basement membrane,GBM) 疾病[1]。自1927年Alpor报道了一个伴神经性耳聋的家系后,该病逐渐受到重视。Alport综合征并非罕见,尤其近10年来随着肾病基因诊断技术的提高及对该病的重视,临床报道逐渐增多,该病的基因研究进展迅速,本文对Alport综合征基因学发病机制研究进展作一综述。
1 AS的分类
按照AS的遗传方式,可以分为伴X染色体显性遗传、常染色体显性遗传和常染色体隐性遗传3种。Mazzucco等[2]对97个家庭108例AS患者进行了基因学及超微结构的研究,其中64个家庭(75例)是X连锁遗传,7个为常染色体隐性遗传,2个常染色体显性遗传,5个无法解释,19个是散发的。X连锁患者约占总AS人数的85%。
2 关于伴X染色体显性遗传的AS
伴X染色体显性遗传的AS病变基因位于X染色体长臂中部Xq21-q22,其基因突变为编码Ⅳ型胶原α5链的COL4A5基因[3] 。COL4A5基因共有51个外显子,大小为140kb,目前发现的该病中COL4A5基因的突变已达300多种。Hertz等[4]对42个该病患者进行了分子生物学的研究,共发现36个突变,包括16个错义突变、7个移码突变、3个框架缺失、4个无义突变、6个接合位点突变。Pan等[5]通过对20个的伴X染色体显性遗传的AS患者COL4A5基因的51个外显子分析发现了5种新的突变形式,它们分别是1位外显子的无义突变,31和43位外显子的2个错义突变,还有2条1和25位的内含子突变,该突变刚好位于它们各自外显子的3′末端。Arrondel等[6]发现波利尼西亚的AS患者均有一个基础突变,该突变以COL4A5的35位外显子的一系列复制为特征, 导致α5链的长度增加约65%,这种α5链仍然能够参与胶原的合成。该病病情往往女轻男重。Topaloglu等[7]报道了一些病情严重的男性和病情轻重不一的女性,研究共发现1616个G至A的替换突变,导致472位上编码精氨酸的密码子突变为编码氨基乙酸的密码子。欧洲联合会AS协作组针对不同家系不同患者进展至终末期肾病(ESRD)的进程不一这一现象进行了研究,目的是评价该病不同家系间疾病的遗传异质性,研究基因型的不同与ESRD进展的关系。研究发现正是基因型的不同导致了家族间表现不一,而且,某些基因型的不同与男性发生ESRD的高危有关。Nakanishi等[8]报道在伴X染色体显性遗传的AS患者中,男性患者通常为ESRD,而女性患者可表现为无症状性血尿,也可表现为ESRD,女性患者的表现各异可能是由不同的X染色体静止形式有关。他们对来自17个家庭的25名伴X染色体显性遗传的AS女性进行了检测,结果表明有许多种α5链的表达,可能是由不同的静止X 染色体形式引起。从而认为这是女性伴X染色体显性遗传的AS患者病情严重程度不一的原因。但也有人认为伴X染色体显性遗传的AS男性与女性基底膜的Ⅳ型胶原组成不同而导致男女性别临床表现的不同,男性α5链染色阴性,由于α5链的结构异常,不能形成正常的α3、α4、α5三螺旋结构并易于降解,故α3、α4链在基底膜中的染色也是阴性的,而伴X染色体显性遗传的AS女性基底膜染色是间断阳性的。AS患者在基因上有同样的突变,而其临床表现却大不相同,说明其他基因或环境也影响着疾病的发生发展。
常染色体显性或隐性AS通常是由COL4A3、COL4A4基因的改变而致, 常染色体隐性AS(ARAS)以COL4A3基因突变居多。这些基因定位于2q35-q37,编码构成Ⅳ型胶原的α3及α4链[9,10],ARAS目前发现的突变位点达100余个,这些突变均为小的突变,包括甘氨酸取代、缺失突变、插入突变、错义突变、无义突变、剪接位点突变、移码突变等。常染色体显性AS(ADAS)的突变包括甘氨酸取代突变、错义突变、剪接位点突变等。Longo[10]等描述了7个COL4A3的突变,一些复合异质接合体的突变与健康异质接合体亲属的微量血尿有关,异质接合体的突变可能是显性的,因为即使通过测序也没有在第二个等位基因上发现突变。COL4A5基因的改变不仅出现在伴X染色体显性遗传的AS中,Mazzucco等[2]发现在常染色体遗传的AS也存在,在超微结构中可有较为严重的突变,突变与超微结构之间无严格的联系。作者在一个无明显症状的患者中发现了一个重要的重排序列,而且在一个家庭中发现了不同的发病模式。即使存在一个基因突变,超微结构特征与α5链基因的表达产物也没有严格的联系。
4 AS的基因前景
AS为基因病变,其治疗到目前为止仍是困扰临床医师的一个难题,至今国内外尚未发现一种有效的治疗方法,如今更多的研究者将目光集中于AS的基因治疗研究上,然而,期望在人类成功地应用基因治疗Alport综合征,仍需进行广泛的、大量的实验研究。目前,已取得了一些有意义的实验进步,应用肾脏灌注腺病毒载体包埋β2半乳糖基因标记物的方法可获取85%的基因转录入猪的肾小球,通过肾脏灌注已能够高效地把基因标记转录入活性肾小球细胞移换后,异常GBMⅣ型胶原网状物被正常的α(Ⅳ)胶原基因融入活性细胞中从而发挥正常的Ⅳ型胶原蛋白功能,这在理论和实验修正方面均已被认可,在带有Alport综合征的萨摩斯狗模型中这一缺陷病因可以被修正。相信不久,人类AS患者的基因治疗将成为可能,基因治疗将给AS患者带来光明的治疗前景。
:
[1] Hudson BG,Tryggvason K,Sundaramoorthy M,et al.Alport syn-drome,Goodpasture syndrome and type Ⅳcollagen[J].N Engl J Med,2003,348:2543.
[2] Mazzucco G,Barsotti P,Muda AO,et al.Ultrastructural and immuno-histochemical findings in Alport’s syndrome : a study of 108 patients from 97Italianfamilies with particular emphasis on COL4A5 gene mutation correlations[J]. J Am Soc Nephrol,1998,9(6):1023.
[3] Palenzuela L,Callis L,Vilalta R,et al. A new point mutation in the COL4A5 gene described in a Spanish family with X -linked Alport syndrome[J]. Nephron,2002,90(4):455.